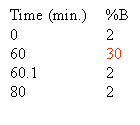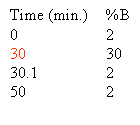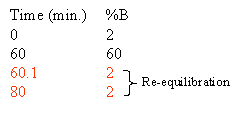Reverse Phase HPLC Basics for LC/MS
An IonSource Tutorial
by
Andrew Guzzetta
This Tutorial was first published
July
22nd, 2001
IonSource
Homepage | Disclaimer
read
important laboratory safety notice at bottom of page before proceeding
We were going to call this tutorial "Reverse Phase HPLC for Proteomics" but we decided to exercise some restraint. We decided to write this tutorial because reverse phase chromatography is the most common form of chromatography used in LC/MS applications. This tutorial is basically targeted to students and those that are new to reverse phase chromatography, HPLC, and LC/MS. The tutorial addresses RP HPLC of peptides and proteins but the principles described can be applied toward the chromatography of any compound.
Table of Contents |
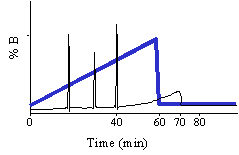
The message of this tutorial is that reverse phase HPLC is simple. Compounds stick to reverse phase HPLC columns in high aqueous mobile phase and are eluted from RP HPLC columns with high organic mobile phase. In RP HPLC compounds are separated based on their hydrophobic character. Peptides can be separated by running a linear gradient of the organic solvent. I often tell my fellow researchers to run the 60/60 gradient when chromatographing an unknown. The 60/60 gradient means that the gradient starts at near 100% aqueous and ramps to 60% organic solvent in 60 minutes. The majority of peptides (10 to 30 amino acid residues in length) will elute by the time the gradient reaches 30% organic. To learn some of the simple principles of RP HPLC please read on.
|
| The HPLC
In most cases the HPLC you intend to use must be able to pump and mix two solvents. This can be accomplished with one pump and a proportioning valve or by using two separate pumps. Generally the pumping configuration is an aspect of the instrumentation that is transparent to the user. Reverse phase chromatography can also be performed in a purely isocratic mode where the solvent conditions are held constant, this form of reverse phase chromatography can be carried out with a single pump. Isocratic methods are used most often in a QC environment in which a single analyte has been extensively characterized and the compound is being run to confirm it's identity and to look for closely related degradation products. If you do not own an HPLC here is a link to HPLC vendors and accessory suppliers.
HPLC Column Components and Specifications
The reverse phase solvents are by convention installed on the HPLC channels A and B. The A solvent by convention is the aqueous solvent (water) and the B solvent by convention is the organic solvent (acetonitrile, methanol, propanol). It is important to follow this convention since the terms A and B are commonly used to refer to the aqueous and organic solvents respectively. The A solvent is generally HPLC grade water with 0.1% acid. The B solvent is generally an HPLC grade organic solvent such as acetonitrile or methanol with 0.1% acid. The acid is used to the improve the chromatographic peak shape and to provide a source of protons in reverse phase LC/MS. The acids most commonly used are formic acid, triflouroacetic acid, and acetic acid. A 0.1% v/v solution is made by adding 1ml of acid per liter of solvent. Triflouroacetic acid has been reported to suppress MS ionization and often mass spectroscopists lower the percentage of TFA to 0.05 or even 0.02% without significant loss in chromatographic efficiency. Some MS people add a small percentage of heptafluorobutyric acid (HFBA, pdf from Michrom) to acetic acid solvents or low TFA containing solvents to help improve peak shape. Since modern mass spectrometers are very sensitive it is important not to use plastic pipette tips when adding acid to the mobile phase, always use glass. In our work we use acetonitrile as our organic solvent. We have heard that the best electrospray solvent is 30% methanol, 35 mM acetic acid. We commonly use this solvent system for ESI MS infusion, but have found that acetic acid is an inferior acid for chromatographic peak shape. Our preferred HPLC grade water, acetonitrile and methanol is purchased from Burdick and Jackson. Our preferred TFA comes in 1 ml ampoules from from Pierce Chemical Company.
When chromatographing an unknown we normally use the following simple gradient to learn about the hydrophobic character of the unknown compound. The % A in the gradient described below is implied. We call this the 60/60 gradient, because we run from near 0% B to 60% B in 60 minutes. Through experience we have noted that 90% of all peptides will elute from a C18 reverse phase column by 30% acetonitrile. There may be a few really hydrophobic peptides that elute later that is why we take the gradient to 60% B. You may even want to run this gradient to 80% at least once to see if you are getting everything off of the column. You may ask why don't we start the gradient at 0% B? As we talked about before, in 0% organic and in high aqueous, the very hydrophobic, long C18 alkyl chains in an effort to get away from the high aqueous environment mat down on the particle. When these alkyl chains mat down they are inefficient at capturing the analyte so chromatographers in the know start the gradient with some small % of organic, 2-5%.
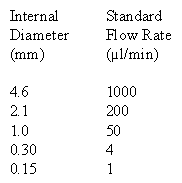
It is important to use the correct flow rate for your HPLC column. Below is a table with standard flow rates for easy reference. If you are running a column with a different diameter than those shown in the table please review the maintaining linear velocity page to learn how to calculate the appropriate flow rate for your column.
Based on the separation shown at the top of this section one could rewrite the gradient to look like this:
The column must be equilibrated, re-equilibrated to the initial high aqueous solvent composition before another analysis can be performed. Normally this re-equilibration is stuck onto the end of the gradient. How much equilibration time is enough? As a rule of thumb we give 20 minutes. In reality it depends on the column length, flow rate and the hydrophobicity of your peptides. Some chromatographers use 10 minutes as their standard equilibration time. Equilibration is all about fitness of purpose. You should determine the the equilibration time experimentally, the criteria will be, does my analyte really stick to the column and chromatograph appropriately and reproducibly with subsequent analyses. If you choose to do this part of the method development you will undoubtedly be rewarded with improved chromatography and better cycle time.
Should I Control Column Temperature? Yes. Scientists are control freaks. If you can control a variable, control it! Actually if you are performing automated analyses over a long period of time peak retention times can drift with changing ambient temperature. It is common for many companies and institutions shut down the air conditioning at night to save money, which could result in shifting peak retention times due to dramatic changes in ambient temperature. Many HPLCs provide the option to control column compartment temperature. If your HPLC does not have this capability a heated column jacket can be purchased from many suppliers. The most common running temperature is 40C, this places the column compartment well above even the most extreme ambient temperature fluctuations. In addition to maintaining constant temperature, temperature can be used to influence the chromatographic separation. No chromatographic study is complete without a temperature study. In our experience higher temperature is better, peaks will be sharper and elute earlier. It is not too uncommon to perform chromatography at 60 C and some daredevils even go to 80 C. Remember though that higher temperature will lead to a shorter column lifetime and some columns may not be able to tolerate 60 C. Consult the manufactures recommendations when experimenting with high temperature. After your runs are complete for the day it is advised that you turn off your column heater since high temperature leads to stationary phase deterioration.
Preparing for the First Run of the Day One observation is that if you start up a reverse phase analysis from a dead stop with a column that has perhaps been sitting in high aqueous conditions for up to 10 hours the analysis will give irreproducible results. Conventional wisdom has it, you want to first flush the column with the highest % organic of your method for at least 3 column volumes and then bring it back to the equilibrating condition. This practice may have the advantage of getting you to standard equilibration conditions faster and it will also clean your column. A better alternative is to make the first run a blank run (or "preparation run") and then the next run can be your real analysis. We prefer the second option because it should get you to the standard starting conditions more accurately. However, often, if we are in a hurry and the first option is quicker, well.....
We store our columns in 50/50 methanol/water without any acid. If you are using a salt, unlikely in LC/MS, wash your entire system, solvent bottles, HPLC, solvent lines, and column, into a non-salt containing solvent. Salt may precipitate out and plug your HPLC or column or may cause corrosion. Usually we flush with pure water first then leave the system in 50/50 methanol: water. Some salts may precipitate out in high organic so an initial water wash is advised. The 50/50 methanol:water solution helps to stop bacterial growth which can muck up your system. Take care of your HPLC, it's the right thing to do!
How Much Protein Should I Load?
|
| Links to Related IonSource Material |
| Calculating
the Appropriate Flow Rates for Columns of Differing Diameters, Maintaining
Linear Velocity
HPLC
Tubing Volume Calculator |
| Links
to External HPLC Information
Journal of Chromatographic Science Quality HPLC Links from LCResources Vydac's, "The Handbook of Analysis and Purification of Peptides and Proteins by Reversed-Phase HPLC"
|
|
|
| Important
Safety Information: Triflouroacetic acid, formic acid,
heptaflouobutyric acid and acetic acid are all very caustic reagents.
Acetonitrile, methanol, and propanol are harmful solvents Consult the material safety data sheets
(MSDS) that come with these reagents and get
the permission of the safety officer at you company or institution
before performing these experiments. Always wear the appropriate safety
apparel; safety glasses, lab coat, and gloves. Use a fume hood when
appropriate. If you are not trained in laboratory safety you should
not attempt these procedures. Read our disclaimer, follow the
link at the bottom of this page.
|
|
home | disclaimer visitors |