
`
Proteomics Protein Identification Philosophy
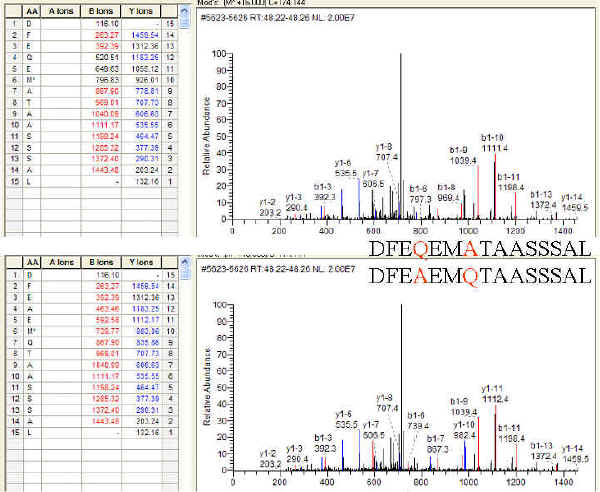
With mass spectrometry data we are at the mercy of the sequence database. If the protein sequence is not entered in the database then we obviously will not be able to identify it with MS matching techniques. Protein databases are constructed by humans, and sadly not by divine inspiration. Researchers make contributions to public databases usually through protein Edman sequencing efforts made in their research projects. Others sequences are garnered directly from publications, and patents. These sequences are prone to a number of errors: human sequencing errors, human transcriptional errors, typographical errors, and database curation errors. In addition it is difficult to know the theoretical absolute chemical composition of large proteins due to a myriad of modifications that occur within these proteins. Degradations are common, oxidation of methionine, deamidation of asparagine and glutamine, backbone cleavages and a myriad of other amino acid modifications can and do occur. In addition post translational modifications are common, for example, glycosylations and phosphorylations which can shift both mass and charge. For a larger more complete list of modifications the reader is referred to UNIMOD, "A Database of Protein Modifications."
The typical "good" MS based protein hit will correlate a
protein with about 30% of sequence covered perhaps with 7 peptides
identified. If you take a look at some of the proteomics studies
currently being published, proteins are being identified with one or two
peptides. This is most definitely a homology identification.
In the strictest sense it is a homology identification at the peptide level and by
definition a homology identification at the protein level. To get
closer to an absolute identification one would need an accurate mass
measurement of the intact protein to match to the peptide correlated
database hit. We are really identifying
correlating peptides. This is not an identification it is a
correlation.
Discouraged? Don't be. The protein, proteomics universe is a wild place, and while it is still wild there is much opportunity to be had. Will you be the next proteomics genius? Maybe. Read the next page to learn what we do to be more certain.

Copyright � 2004-2016 IonSource All rights reserved.
Last updated: Tuesday, January 19, 2016 02:48:32 PM