
Searching A Sequence Database With Complex Proteomic Data
So far we have learned how to identify proteins with peptide-mass fingerprinting, sequence tag, and with un-interpreted MS/MS data. Most of the procedures we have performed have been fairly manual. Generally proteomic data is gathered during an LC/MS run, that is, mass spectra are collected during an HPLC separation of a complex mixture of peptides. Modern mass spectrometers can collect 3 to10 fragment spectra per second. Most proteomic LC/MS analyses last several hours, and most multidimensional separations, i.e. strong cation exchange followed by reverse phase LC/MS, can take anywhere from 12 to 24 hours, and that is for a single sample. A two hour run with a mass spectrometer collecting 10 fragment spectra every second could yield a lot of spectra. If you don't have a hundred graduate students, then sophisticated software is your only option.
A Complex Proteomic Protein ID Exercise:
Use the short but complex LC/MS run shown in Figure 1 below, and search a sequence database. For this example you will be using the web version of !Xtandem, part of "The Global Proteome Machine" at UBC
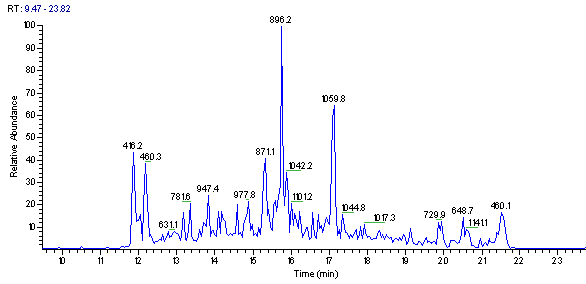
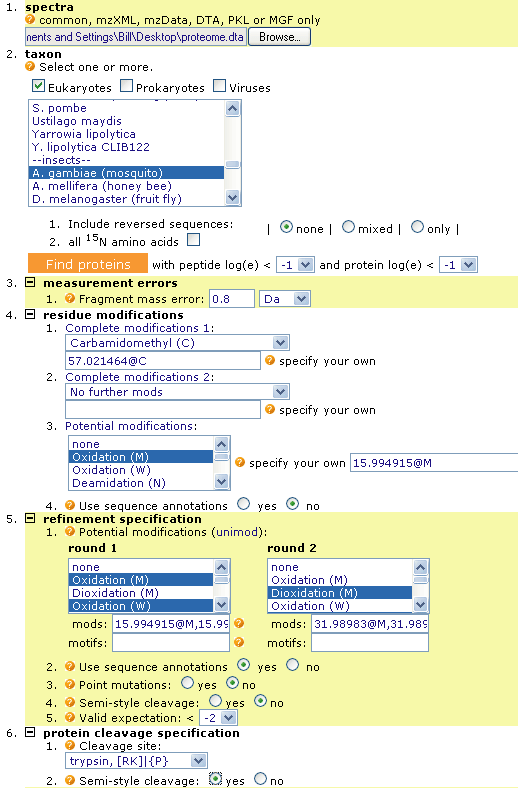

-
Once you are finished making the changes to the submission form Press the Find Proteins button near the top third of the page, it is an orange button. The search will take a few minutes, there are many MS/MS spectra contained in the file you just submitted.
Suggested Reading For This Module:
-
Olsen JV, Mann M. Improved peptide identification in proteomics by two consecutive stages of mass spectrometric fragmentation. Proc Natl Acad Sci U S A. 2004 Sep 14;101(37):13417-22. Epub 2004 Sep 03. PMID: 15347803
- -
Colinge J, Masselot A, Cusin I, Mah� E, Niknejad A, Argoud-Puy G, Reffas S, Bederr N, Gleizes A, Rey P-A, Bougueleret L. "High performance peptide identification by tandem mass spectrometry allows reliable automatic data processing in proteomics," Proteomics. 2004 Jul;4(7):1977-84.
- -
Colinge J, Masselot A. "Mass spectrometry has married statistics: uncle is functionality, children are selectivity and sensitivity," DDT: TARGETS, Vol. 3, No. 2 (Suppl.), 2004, pp. 50-55.
- -
Magnin J, Masselot A, Menzel C, Colinge J. OLAV-PMF: a novel scoring scheme for high-throughput peptide mass fingerprinting. J Proteome Res. 2004 Jan-Feb;3(1):55-60. PMID 14998163
- -
Colinge J, Magnin J, Masselot A. A systematic statistical analysis of ion trap tandem mass spectra in view of peptide scoring. In: Proceeding of the Workshop on Algorithms in Bioinformatics (WABI), R. Page and G. Benson (Eds), Budapest, September 2003, LNBI 2812, Springer, 25-38, 2003.
- -
Colinge J, Masselot A, Giron M, Dessingy T, Magnin J.. "OLAV: towards high-throughput tandem mass spectrometry data identification," Proteomics, Vol. 3, No. 8, August 2003, pp. 1454-1463.
- - McDonald
WH, Yates JR 3rd. Shotgun proteomics: integrating technologies to
answer biological questions. Curr Opin Mol Ther. 2003
Jun;5(3):302-9. Review. PMID: 12870441
- -
Colinge J, Magnin J, Dessingy T, Giron M, Masselot A. Improved peptide charge state assignment. Proteomics. 2003 Aug;3(8):1434-40. PMID: 12923768
- -
Lin D, Tabb DL, Yates JR 3rd. Large-scale protein identification using mass spectrometry.
Biochim Biophys Acta. 2003 Mar 21;1646(1-2):1-10. Review.PMID: 12637006 -
Hunter TC, Andon NL, Koller A, Yates JR, Haynes PA. The functional proteomics toolbox: methods and applications. J Chromatogr B Analyt Technol Biomed Life Sci. 2002 Dec 25;782(1-2):165-81. Review. No abstract available.PMID: 12458005
-
McDonald WH, Yates JR 3rd. Shotgun proteomics and biomarker discovery.
Dis Markers. 2002;18(2):99-105. Review. PMID: 12364816
 -
-
Copyright � 2004-2016 IonSource All rights reserved.
Last updated: Tuesday, January 19, 2016 02:48:31 PM