
Protein ID With MS/MS Data
Step 1. Step one usually begins with
placing an enzyme specificity constraint. Some programs pre-index the sequence
database based on the enzyme specificity. This makes the search go
much faster as for example tryptic peptides can be indexed and mass lists
can be pre-made. The downside is a separate database needs to be indexed for each
enzyme or each time a potential modification is changed.
-
Step 2. Step two involves matching the
parent mass of the intact peptide to the peptides in the database. Generally
the narrower you can set the parent mass constraint the faster the search
will go because fewer peptides will need to be correlated in the next step. One needs to be certain
that the accuracy of the mass spectrometer is not exceeded when setting the
parent mass window in the search. In this step a short
list of peptides are selected for the fragment mass correlation in the next step.
-
Step 3. Step three takes the list of
peptides identified by parent mass in step two and compares the theoretical fragment
masses of these peptides to the experimentally derived fragment spectra.
-
Step 4. Peptide Ranking. Hits are ranked by
how many of the fragment masses match the theoretical fragment masses in the
sequence database.
Step 5. If more than one peptide is
searched all of the peptides found are correlated to their prospective
proteins. The protein with the greatest number of well correlated
peptides is usually the most significant hit.
-
Step 6. Some programs go a bit further using probability to
back-up the proposed match. Most users like to have a real probability
number, this gives them some comfort in the designation. Probability
takes the onus off of the user. While probability comforts us, we find
for the most important individual protein hit we still go in and manually
validate the spectral match.
Simple MS/MS Protein ID Exercise:
Use the MS/MS spectrum in Figure1 below to search a sequence database. For this example we will be using the web version of the database searching program Phenyx, this program is made available by GeneBio.
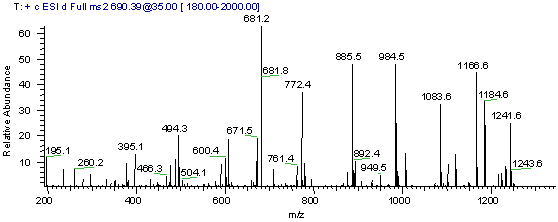
Exercise Procedure:
You can use the dta file
that we have already created for you, right
click on this dta link,
and save this file to your computer, choose save target as. When
saving the file to your computer, during the saving dialog you may
need to select "all files" as the file type and you may also need
to add the suffix .dta to
the "msms" name, in order for your computer to get the file type
correct. A dta file
is a mass / intensity pair list that is a representation of the original
MS/MS spectrum, the dta file is a Thermo Finnigan file format. The dta file
is very small, a few KB, once you have it downloaded, if you want, you can
open it with notepad to inspect the simple file format.
Steps to follow on the Phenyx submission page:
1.) Follow this link
to the Phenyx web portal.
2.) Click on the "Log In"
button on the Phenyx page to log in as a guest. (We recommend
that you create your own account, so that you can keep your future data
submissions private and so that you can keep track of past and future submissions.)
3.) Then click on the Submission
button. The main submission page will come up, do the following:
A.) Change taxonomy to "root."
You will need to scroll up to the top of the list to find root.
(root means that the program will search all species.)
B.) Change the algorithm to LCQ.
(this is the instrument we used)
C.) Click on the "Browse"
button and browse to and load the msms.dta file that you just downloaded.
D.) Look for the file format
field, and change the file format to dta.
E.) Click on the Submit
button.
Wait a few seconds until the "link to result" says, "available",
then click on the link below it. If you do not have pop ups blocked,
like we do, the results page would have appeared
automatically.
Inspect the results page:
Conclusion:
Mass spectrometry based protein identification is too easy right? So far we have used three MS sequence database searching techniques to identify a single protein. Continue on to the final tutorial to learn how to identify many proteins in a somewhat typical proteomics experiment.
References:
-
Jimmy K. Eng, Ashley L. McCormack and John R. Yates, III An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database JASMS, Volume 5, Issue 11,
November 1994 ,Pages 976-989 -
Yates JR 3rd, Eng JK, McCormack AL, Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database.
Anal Chem. 1995 Apr 15;67(8):1426-36. PMID: 7741214 -
Yates JR 3rd, Eng JK, McCormack AL. Mining genomes: correlating tandem mass spectra of modified and unmodified peptides to sequences in nucleotide databases.
Anal Chem. 1995 Sep 15;67(18):3202-10. PMID: 8686885 -
Lim H, Eng J, Yates JR 3rd, Tollaksen SL, Giometti CS, Holden JF, Adams MW, Reich CI, Olsen GJ, Hays LG. Identification of 2D-gel proteins: a comparison of MALDI/TOF peptide mass mapping to mu LC-ESI tandem mass spectrometry. J Am Soc Mass Spectrom. 2003 Sep;14(9):957-70. PMID: 12954164

Copyright © 2004-2016 IonSource All rights reserved.
Last updated: Tuesday, January 19, 2016 02:48:20 PM