
`
Peptide Sequence Tag
ESI Ion Trap Generated MS/MS Mass Spectrum
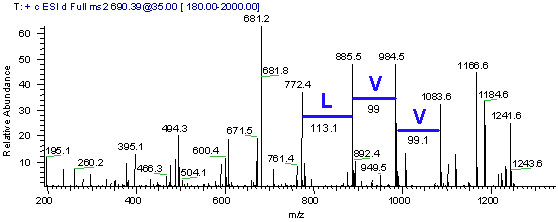
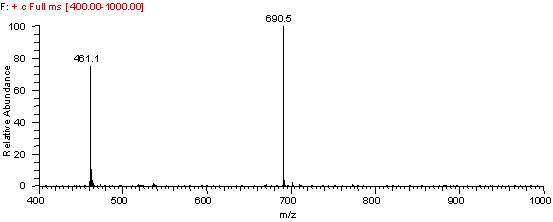
As a component of the search the neutral peptide mass must be determined from the regular MS spectrum. For a description on determining the neutral mass of a peptide please see our tutorial on ESI mass spectrum interpretation. Take a look at the full mass spectrum used to select the mass for MS/MS fragmentation, see Figure 2. Use the spectrum in Figure 2 to determine the neutral mass of the peptide. Remember these two peaks are related mathematically. The x-axis is mass/charge.
Full ESI Ion Trap Mass Spectrum
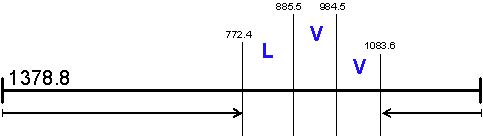
The Ultimate Constraint:
The Ultimate Flexibility: This approach also allows for homology matching when the program is asked to match only two of the three regions. In this way one of the terminal regions is allowed to float. The sequence tag is maintained and the program only needs to match the mass difference before or after the tag. While you lose some of the power of the original constraint this can be a way to search when the traditional sequence tag fails to find a match due to peptide modification.
Conclusion: Most mass specromitrists have fond memories of this technique since it was one of the first freely available programs, and helped many scientist whose bioinformatics resources were limited. The original program does suffer from some surmountable problems: the manual nature of calling the sequence tag, the manual nature of determining the neutral peptide mass, and the problem of not knowing whether one is calling a "b" or "y" ion series. Hence, as we have entered the era of high through-put proteomics style peptide sequencing and identification this technique has suffered a decline in popularity. One can envision a rebirth and dominance of this technique once an automated de-novo sequencing program is able to call the sequence tag automatically with high probability. Again the manual nature of this technique has slowed it�s popularity in recent years.
- Mann M, Wilm M., Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal Chem. 1994 Dec 15;66(24):4390-9.
- Mortz E, O'Connor PB, Roepstorff P, Kelleher NL, Wood TD, McLafferty FW, Mann M. Sequence tag identification of intact proteins by matching tanden mass spectral data against sequence data bases. Proc Natl Acad Sci U S A. 1996 Aug 6;93(16):8264-7.PMID: 8710858
- Shevchenko
A, Jensen ON, Podtelejnikov AV, Sagliocco F, Wilm M, Vorm O, Mortensen
P, Shevchenko A, Boucherie H, Mann M. Linking genome and
proteome by mass spectrometry: large-scale identification of yeast
proteins from two dimensional gels.
Proc Natl Acad Sci U S A. 1996 Dec 10;93(25):14440-5. PMID: 8962070
|
|
 |
|
Copyright � 2004-2016 IonSource All rights reserved.
Last updated: Tuesday, January 19, 2016 02:48:33 PM